大疱性表皮松解症
痒疹型大疱性表皮松解症(EpidermolysisBullosaPruriginosa,EBP)是营养不良型大疱性表皮松解症(DystrophicEpidermolysisBullosa,DEB)的一种罕见临床亚型,故又称痒疹型营养不良型大疱性表皮松解症最早于1994年由McGrath等人首报。
大疱性表皮松解症(EB,Epidermolysisbullosa)是一种罕见的遗传疾病,表现为皮肤非常脆弱,因日常的轻微摩擦而反复发作水疱,大约5万个新生儿中会有一个患EB,所有种族都会患EB,并且男女比例相同。
大疱性表皮松解症的形成原因
大疱性表皮松解症[1]可呈常染色体显性或隐性遗传,主要由编码皮肤结构蛋白的基因突变所致,目前已发现的致病基因达20个,不同类型的遗传性大疱性表皮松解症的致病基因不同。
基本原因
- 单纯型EB:大部分为常染色体显性遗传,由KRT5和KRT14基因发生突变,基因发生突变后无法编码表皮基底层的角蛋白,引起细胞骨架严重破坏,导致皮肤脆性增加,PLEC1、桥粒斑蛋白基因发生突变也可导致单纯型EB;
- 交界型EB:与COL17A1、ITGB4和ITGA6基因发生突变相关;
- 营养不良型EB:可为染色体显性遗传或隐性遗传,是由COL7A1基因发生突变所致。
EBS的基因突变发生在角蛋白基因K5和K14上,DEB的基因突变发生在角蛋白COL7A1上,两者基因位点不同,因此虽然表现症状都为水疱,但水疱部位各不相同。
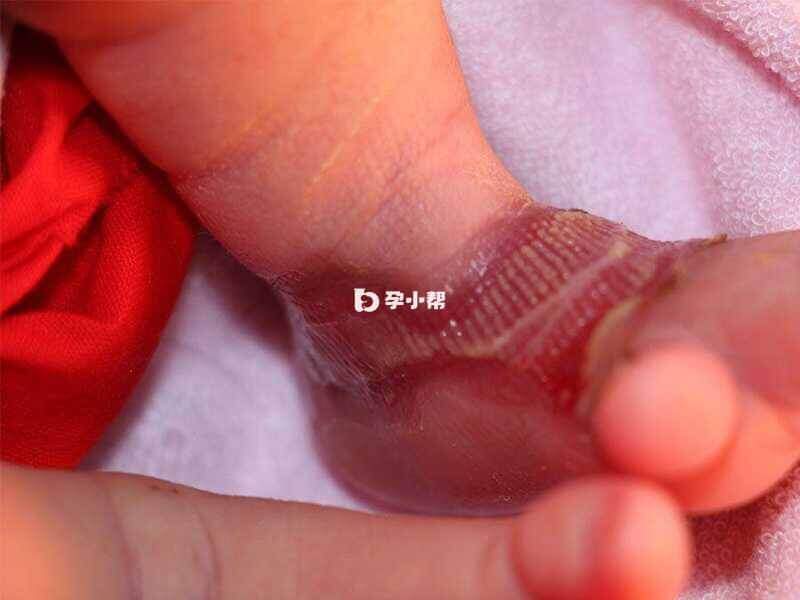
大疱性表皮松解症的临床表现
临床表现为身体容易摩擦的部位,如手部、肘关节、膝关节及足部,甚至全身出现水疱或大疱,可有糜烂或溃疡,口腔咽喉部黏膜发生水疱或溃疡,会导致声音嘶哑、吞咽困难,水疱愈合后,可有皮肤色素异常、瘢痕或皮肤萎缩,本病患者皮损及皮损愈合部位发生皮肤癌的概率也相应增高。
具体表现
- 1. 单纯型(EBS):症状最轻,孩子出生后几个月,刚学会爬行时手、四肢关节伸侧开始出现水疱,随着孩子长大,青春期后可能痊愈;
- 2. 营养不良型(DEB):临床上可分为显性和隐性遗传两种,后者病变广泛而严重,手指或者口腔都会受到感染,患病的小孩可因发育受阻而导致死亡。而显性遗传者则相对比较轻;
- 3. 交界型(JEB):最为罕见而且最危险的一种,新生儿在出生的那刻即可看到有严重广泛性分布的大疱和大面积的表皮剥脱,可于数日至数月内死亡,婴儿如幸存,其后仍可发生生长迟缓,以及中或重度的顽固性贫血,多数患儿于2岁内死亡。

严重的EB患者的水疱不局限于体表皮肤,体内的软组织(粘膜)也会有水疱,包括口腔、食道、胃、肠、肺、膀胱和尿道,体内水疱的范围每个人都不同,取决于所患EB的亚型和疾病程度。
大疱性表皮松解症的治疗方法
尽管EB无法彻底治愈,但很多只要积极治疗就可以缓解痛苦,随着年龄的增长,症状会逐步减轻,到后期,只需要很少治疗或完全不需要治疗,最主要的方法为:防止感染,保护皮肤免受摩擦,如有摩擦引起的水疱,需要及时用碘酒清洗消毒,如果有严重水疱患者,则建议送医院治疗。
瘙痒程度严重者,可适当服用抗过敏药物,防止搔抓造成皮肤进一步损伤,常用药物有盐酸西替利嗪片等,但要注意可能会出现头痛、嗜睡、口干等副作用,对盐酸西替利嗪过敏、妊娠及哺乳期妇女禁用盐酸西替利嗪,可外用莫匹罗星软膏预防或治疗继发感染,偶尔会出现局部灼热、瘙痒或过敏等不良反应。
孕小帮上所有内容均出于传递更多信息之目的,并不意味着赞同其观点或证实其描述。
任何关于疾病的建议都不能替代执业医师的面对面诊断,请谨慎参阅。本站不承担由此引起的法律责任。